可变多聚腺苷酸化(Alternative polyadenylation)
workflow
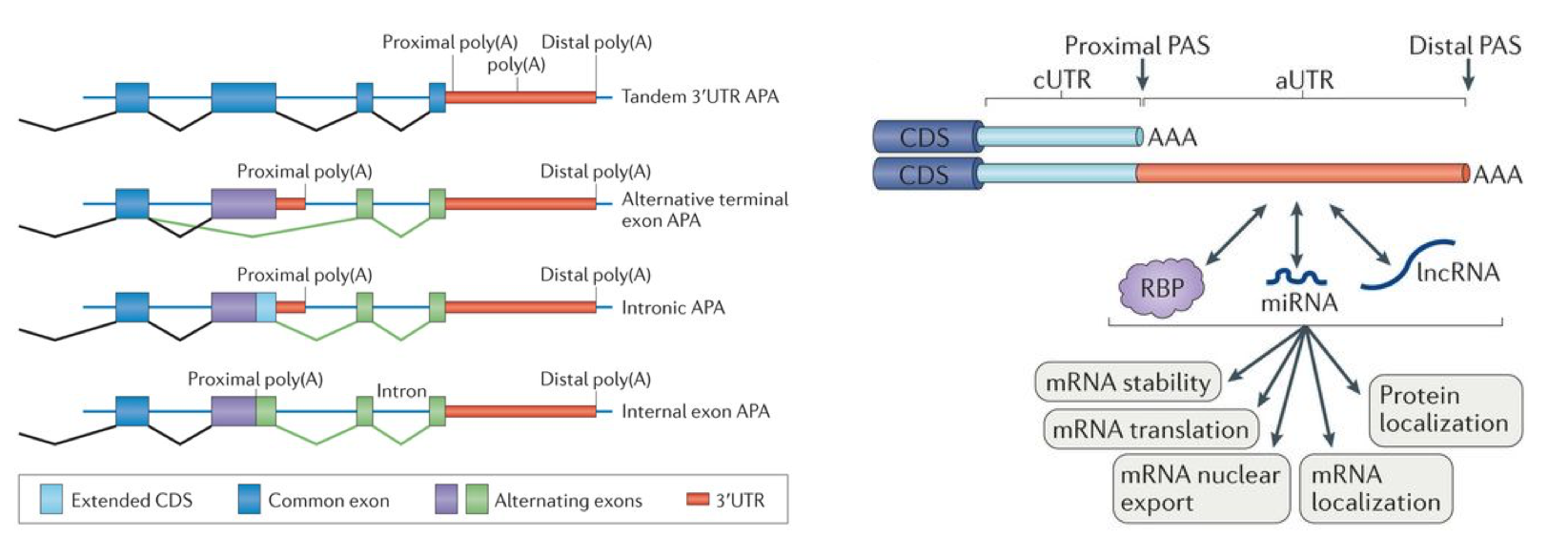
Background
Alternative polyadenylation (APA) leading to the production of two mRNA isoforms with different 3ʹ untranslated regions (3ʹ UTRs)The dynamic usage of the 3’untranslated region (3’UTR) resulting from alternative polyadenylation (APA) is emerging as a pervasive mechanism for regulating mRNA diversity, stability and translation.
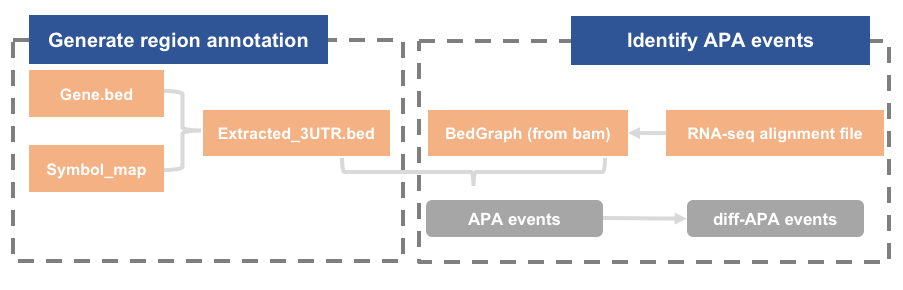
Data Processing
DaPars
step1. Generate region annotation: python DaPars_Extract_Anno.py -b gene.bed -s symbol_map.txt -o extracted_3UTR.bed
DaPars will use the extracted distal polyadenylation sites to infer the proximal polyadenylation sites based on the alignment wiggle files of two samples. The output in this step will be used by the next step.
python DaPars_Extract_Anno.py -b hg19_refseq_whole_gene.bed -s hg19_4_19_2012_Refseq_id_from_UCSC.txt -o hg19_refseq_extracted_3UTR.bed
input
- hg19_refseq_whole_gene.bed (bed12 format)
chr1 66999824 67210768 NM_032291 0 + 67000041 67208778 25 227,64,25,72,57,55,176,12,12,25,52,86,93,75,501,128,127,60,112,156,133,203,65,165,2013, 0,91705,98928,101802,105635,108668,109402,126371,133388,136853,137802,139139,142862,145536,147727,155006,156048,161292,185152,195122,199606,205193,206516,207130,208931, chr1 33546713 33585995 NM_052998 0 + 33547850 33585783 12 182,121,212,177,174,173,135,166,163,113,215,351, 0,275,488,1065,2841,10937,12169,13435,15594,16954,36789,38931, chr1 16767166 16786584 NM_001145278 0 + 16767256 16785385 104,101,105,82,109,178,76,1248, 0,2960,7198,7388,8421,11166,15146,18170, - hg19_4_19_2012_Refseq_id_from_UCSC.txt
#name name2 NM_032291 SGIP1 NM_052998 ADCoutput
hg19_refseq_extracted_3UTR.bed
chr14 50792327 50792946 NM_001003805|ATP5S|chr14|+ 0 + chr9 95473645 95477745 NM_001003800|BICD2|chr9|- 0 - chr11 92623657 92629635 NM_001008781|FAT3|chr11|+ 0 +
step2. Main function to get final result
python DaPars_main.py configure_file
Run this function to get the final result. The configure file is the only parameter for DaPars_main.py, which stores all the parameters.
input
- configure_file
The format of the configure is:
#The following file is the result of step 1.
Annotated_3UTR=hg19_refseq_extracted_3UTR.bed
#A comma-separated list of BedGraph files of samples from condition 1
Group1_Tophat_aligned_Wig=Condition_A_chrX.wig
#Group1_Tophat_aligned_Wig=Condition_A_chrX_r1.wig,Condition_A_chrX_r2.wig if multiple files in one group
#A comma-separated list of BedGraph files of samples from condition 2
Group2_Tophat_aligned_Wig=Condition_B_chrX.wig
Output_directory=DaPars_Test_data/
Output_result_file=DaPars_Test_data
#At least how many samples passing the coverage threshold in two conditions
Num_least_in_group1=1
Num_least_in_group2=1
Coverage_cutoff=30
#Cutoff for FDR of P-values from Fisher exact test.
FDR_cutoff=0.05
PDUI_cutoff=0.5
Fold_change_cutoff=0.59
output

step3. Filter diff-APA events
FDR_cutoff, PDUI_cutoff, Fold_change_cutoff → Pass filer (Y nor N)